Chapter 15: HAEMATOLOGICAL AND ONCOLOGICAL CONDITIONS
Student Authors: Neethu Benny Varghese and Nazneen Pilodia
Specialist Advisor: Dr Helder de Quintal

This chapter covers the following topics:
- Anaemia
- Idiopathic thrombocytopenic purpura (ITP)
- Haemophilia
- Disseminated intravascular coagulopathy (DIC)
- Von Willebrand disease (vWD)
- Oncology
- Leukaemia
- Lymphoma
- Wilms Tumor (Neuroblastoma)
- Neuroblastoma
- Retinoblastoma
- Rhabdomyosarcoma
Anaemia
It is defined as haemoglobin (Hb) or haematocrit (Hct) below the normal value for age and sex. The cut-off values for anaemia for the different age groups are shown in table 15.1 and are based in WHO/Integrated Management of Childhood Illness (IMCI) guidelines).
Table 15.1: Anaemia Cut-off Values
| Age (years) | Hb (g/dl) |
|---|---|
| 0.5 - 4.9 | < 11 |
| 5 - 11.9 | <11.5 |
| 12 - 14.9 | <12 |
Severe anaemia is diagnosed in the child with Hb <5 g/dl.
Iron-Deficiency Anaemia (IDA)
Iron deficiency is the most common cause of anaemia in early childhood, ranging in prevalence from 5% in Western societies to 50% in developing countries. It develops when body iron stores are too low to support erythrocyte production. Term neonates have enough iron reserves for 3 months, after which they get iron from their food. This is not so for preterm neonates. Thus, iron supplementation is required from birth weight for these neonates.
Aetiology and Predisposing Factors
Iron deficiency may be caused by:
- Inadequate dietary intake
- Increased requirements (as in infancy, adolescence and pregnancy)
- Impaired absorption e.g. gluten enteropathy, H. pylori infection
- Blood loss e.g. menses in females, GIT bleeding (as with helminthiasis, peptic ulcers, diverticulosis or colonic malignancy)
- Chronic loss of body iron in urine
Bacterial, viral and parasitic infections impair iron uptake and utilisation or cause chronic blood loss.
Prenatal predisposing factors (factors which result in depleted iron stores at birth) for IDA include:
- Maternal multiparity
- Multifoetal pregnancies e.g. twin births
- Low birth weight
- Blood loss
Clinical Features
They include:
- Initial symptoms:
- Fatigue and lethargy (caused by depletion of tissue enzyme and myoglobin iron)
- Irritability
- Anorexia or pica
- Long-term manifestations:
- Cognitive dysfunction
- Growth impairment
- Impaired immunity
- Examination findings:
- Pallor
- Koilonychia
- Angular stomatitis
- Glossitis
- Moderate splenomegaly
- Soft ejection systolic murmur
Assessment and Investigations
When one suspects IDA, one must ask the mother about the factors and causes listed above. The following blood results will be found in the child with IDA:
- Low serum iron (<40 µg/dl)
- High iron binding capacity (>450 µg/dl)
- Low ferritin (due to the low serum iron and transferrin saturation)
- Low transferrin saturation (<16%)
- Low serum ferritin (<10ng/mL)
- High free erythrocyte protoporphyrin level (FEP)
- High RDW
- Microcytosis (mean corpuscular volume; MCV <70 fL)
- Hypochromia (mean corpuscular Hb; MCH <26 pg)
- Pencil cells, target cells and reactive thrombocytosis on the peripheral blood smear (PBS)
The reticulocyte count will be low for the degree of anaemia, unless the anaemia is caused by acute blood loss.
Measurement of bone marrow iron is the gold standard for assessing iron stores. One will see reduced iron stores on Perl’s Prussian blue stain. However, it is an invasive test and is, thus, not routinely done.
- To determine another cause of microcytic anaemia:
- stool testing
- incubated osmotic fragility testing
- measurement of lead in tissue
Diagnosis and Differential Diagnosis
IDA is diagnosed based on the presence of supporting clinical features and investigations. Response to iron supplementation (improvement of lethargy and fatigue) usually confirms the diagnosis. Differential diagnoses for the hypochromic microcytic anaemia in children aged 6 months to 3 years are:
- Thalassaemia
- Chronic infections or inflammatory conditions
- Lead poisoning
Management
One must identify and treat the cause, and correct the deficiency with oral or parenteral supplementation. Attempts to prevent iron deficiency in the at-risk child must be made, including:
- Encouraging breastfeeding
- Encouraging the intake of citrus fruits (only if >6 months) as they increase the availability of dietary iron
- Giving supplemental iron (may be given in the form of iron-fortified food) Pharmacological management includes:
- Oral ferrous sulphate supplementation:
- A dose of 6 mg elemental Fe2+/kg/day will correct nutritional iron deficiency in 4 weeks (in >90% of patients).
- The minimum acceptable rise in Hb is 2 g/dl in 3 weeks.
- Parenteral iron:
- It is usually reserved for genuine oral intolerance e.g. gastrointestinal disease, malabsorptive state.
- Blood transfusion:
- It is only indicated in the patient with very severe anaemia (Hb <4 g/dl) and cardiopulmonary symptoms, continuing blood loss or severe infection.
- 5-10 mL/kg of packed red blood cells (RBCs) is given slowly with a diuretic.
Haemolytic Anaemia
It is anaemia which results from:
- Increased RBC destruction (intravascular haemolysis)
- There is a compensatory increase in bone marrow extramedullary (liver and spleen) erythropoiesis.
- Excessive removal of RBCs by the macrophages within the reticuloendothelial system (RES), mainly the spleen (extravascular haemolysis)
Clinical Features
The patient will have:
- Pale mucous membranes
- Mild, fluctuating jaundice
- Splenomegaly
- Gallstones and cholecystitis (present at younger ages)
- Lower leg ulcers
- Aplastic crises precipitated by parvovirus B19 infection
- Bone marrow hyperplasia
Investigations
Laboratory tests may show:
- Anaemia
- High unconjugated bilirubin levels
- Haemosiderinuria, haemoglobinuria and urobilinogenuria
- Reticulocytosis
- Low serum haptoglobin levels
- Evidence of RBC damage on PBS (spherocytes, fragments, bite cells)
- Heinz bodies (aggregates of denatured Hb which are seen in glucose-6-phosphate-dehydrogenase deficiency; G6PD) (see related image here)
Aetiology
The causes of haemolytic anaemia may be hereditary or acquired.
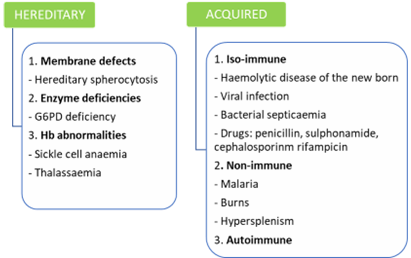
Hereditary Spherocytosis
It is an autosomal dominant condition that is often the result of de novo mutation (25% of cases). The resultant deficiency in spectrin is associated with increased permeability of the cells to sodium. The degree of spectrin deficiency is directly proportional to the severity of disease and degree of spherocytosis. Due to the deficiency, the RBC membrane becomes round and unusually rigid, causing the RBCs to become less pliable. As a result, the cells are unable to pass through the splenic microcirculation and become trapped and destroyed in venous sinuses i.e. the cells’ lifespans are shortened. Thus, a splenectomy will cure the anaemia.
Clinical Features
The clinical presentation in hereditary spherocytosis varies. The patient may be asymptomatic, have compensated haemolysis or have severe recurrent anaemia and require transfusion. The age at presentation is also variable (infancy to old age). Generally, the patient will present with:
- Anaemia with episodes of haemolysis and jaundice
- Splenomegaly (typical)
- Gallstones (in the long term)
Investigations
Laboratory tests will show:
- Spherocytes with polychromasia on PBS (see related image here)
- Reticulocytosis
- Negative Coombs test
- Increased osmotic fragility
Management
Most patients do not require specific therapy and are treated with routine folate supplementation. Patients with severe anaemia may need to be considered for splenectomy, however this does not change the intrinsic cause of the condition. Post-splenectomy risk of pneumococcal septicaemia is decreased when surgery is deferred until the child is older than 5 years, and giving the pneumococcal vaccine and prophylactic penicillin post-operatively.
Hereditary Elliptocytosis
Hereditary elliptocytosis is an autosomal dominant condition in 85-90% of cases and is autosomal recessive in the remaining 10-15% of cases (see related image here). It is characterised by oval-shaped RBCs caused by an abnormality in the RBC skeletal membrane. Patients may be symptomatic or have mild, compensated anaemia. A splenectomy is offered for severe disease.
Glucose-6-Phosphate Dehydrogenase (G6pd) Deficiency
It is most prevalent in people originating from areas with endemic P. falciparummalaria as the deficiency provides limited protection against malaria. The resultant 309 glutathione deficiency makes the RBC susceptible to oxidative stress and shortens its lifespan.
G6PD deficiency is a sex-linked disorder, with the gene being carried on the X chromosome. Thus, full expression of the disease occurs in males and females are carriers (but females are rarely homozygous). Enzyme levels are lower in Caucasian populations than in African populations. Thus, the former may have compensated haemolytic anaemia with increased reticulocytosis and splenomegaly, while the latter display few or no symptoms until exposed to oxidant stress.
Clinical Features and Diagnosis
Neonatal jaundice is a common presenting feature (likely triggered by substances in breast milk). Jaundice and haemoglobinuria are followed by a sharp drop in Hb, brisk reticulocytosis (present on days 3-7) and a return of Hb to normal (in 1-2 weeks). The child may present with:
- Favism (expression of the disease can be triggered by eating fava beans)
- Chronic non-spherocytic haemolytic anaemia
- Drug-induced acute haemolysis
- Neonates may develop haemolysis from
The diagnosis of G6PD deficiency is confirmed by screening tests and a quantitative assay of RBCs.
Management
It includes:
- Avoidance of oxidant drugs
- Blood transfusion in acute haemolytic episodes
- Prompt management of infection
Patients should apply for a MedicAlert bracelet and this should be worn at all times.
Pyruvate Kinase Deficiency
It is an autosomal recessive condition which mainly occurs in northern European people. The diagnosis is confirmed by an enzyme screening test or on RBC assay. Haemolysis may be precipitated by infection.
Glucose-6-Phosphate Isomerase and Hexokinase Deficiency
It is a rare, autosomal recessive condition which is diagnosed on RBC enzyme assay. It is managed with a splenectomy.
Table 15.2: Pathophysiology of Haemoglobinopathies and Thalassaemias
HAEMOGLOBINOPATHIES AND THALASSAEMIAS
Normal Hb is composed of three components: HbA (α2β2 chains), HbA2 (α2δ2 chains) and HbF (α2γ2 chains). HbA accounts for >95% of circulating Hb. HbF is the major Hb type at birth and decreases by 3-4%/week until adult levels are reached (~6 months old).
α-chains are common to all normal Hb. Thus, defects of these chains will manifest before birth. α-chain disorders include Hb Barts, hydrops fetalis, Hb H disease, trait and silent carriers.
Defects of β-chains only manifest at 3-6 months of age when β-chain synthesis occurs. Sickle cell disease is a common β-chain haemoglobinopathy. Other β-chain defects cause less severe haemolysis (Hb C, D and E).
Sickle Cell Disease
This inherited autosomal codominant condition is caused by a substitution of valine for glutamic acid. This renders Hb less soluble on deoxygenation. Tactoids form within the RBCs, distorting their shape, impeding their passage through small capillaries and causing vaso-occlusive crises and haemolytic anaemia. Both parents will show sickle cell trait and patients may be homozygous (HbSS), heterozygous (HbAS) or compound heterozygous (HbSC, HbS/ β-thalassaemia) Sickle cell disease is the most common and most severe inherited disease in Africa. The highest prevalence of the disease is in in West Africa because sickle cell trait offers protection against P. falciparum malaria in endemic areas.
Clinical Features
Intravascular sickling may be caused by acute infections, hypoxia, shock, dehydration, acidosis, or exposure to cold and leads to:
- Painful vaso-occlusive episodes – intense pain in the abdomen, chest or long bones and dactylitis (swelling of hands and feet); may require analgesia (morphine)
- Infarction of the viscera e.g. stroke, acute kidney injury, mesenteric ischemia, acute chest syndrome, splenic auto-infarction with progressive splenic hypofunction and atrophy by about 5 years
- Haemolytic crises with varying degrees of anaemia and jaundice
- Splenic sequestration with severe anaemia and thrombocytopaenia
Other problems may include aplastic crises following infection with parvovirus B19, gallstones and high risk of pneumococcal and haemophilus influenza infection.
Investigations
Laboratory tests will show
- Anaemia (see related image here)
- Sickle cells on blood smear
- Increased osmotic fragility
The diagnosis is confirmed by Hb electrophoresis (will show that the total Hb is comprised of 80% HbS and HbF).
Management
It includes:
- Folate supplementation
- Penicillin VK prophylaxis
- Hydroxyurea 15 mg/kg daily (to stimulate HbF production)
- Prompt treatment of any infections
- Treatment of painful crises with liberal IV or oral fluids and adequate analgesia
- Blood transfusion; considered in the following settings – severe anaemia, splenic sequestration, or aplastic or hyperhaemolytic crises.
Newer treatments include gene therapy and bone marrow transplantation. One should consider enrolling patients with Moya Moya syndrome in a hypertransfusion programme.
In terms of prevention, prenatal testing of parents and/or foetuses in areas with a high prevalence should be offered, as should termination of pregnancy.
Thalassaemia
Thalassaemia is the result of defective mRNA translation and the defective production of mRNA-controlling globin chains, leading to decreased synthesis of α-, β-, δ- or γ-chains. The most common form is β-thalassaemia (no β-chains are synthesised, HbA is absent and HbF predominates). β-thalassaemia is prevalent in people with Middle Eastern and Asian heritage e.g. the Indian community in South Africa. Thalassaemia major (Cooley’s anaemia) is homozygous β-thalassaemia and thalassaemia minor is heterozygous β-thalassaemia (usually asymptomatic), see related image here.
β-thalassaemia is characterised by abnormal haem synthesis, which results in:
- Microcytic, hypochromic erythropoiesis
- Chronic, progressive hemosiderosis
Clinical Features
Patients typically present at 4-6 months with:
- Anaemia – severe, microcytic, hypochromic anaemia with poikilocytes, target cells, stippled cells and increased reticulocytes
- Jaundice
- Fever
- Impaired growth
- Hepatosplenomegaly
Skull bossing and maxillary hypertrophy develop in the first 2 years due to extramedullary erythropoiesis. Progressive haemosiderosis leads to hepatic, cardiac and endocrine dysfunction. The child will develop chronic, transfusion-dependent haemolytic anaemia and experience frequent infections. Few survive beyond a decade without treatment (see related image here).
Investigations
Laboratory tests will show elevated serum iron, transferrin saturation, serum ferritin and bone marrow iron (all due to chronic transfusions). The diagnosis is confirmed with Hb electrophoresis.
Management
The patient is treated with regular transfusions (reduces the phenotype) and oral iron chelators. Desferrioxamine is given overnight by continuous subcutaneous infusion to promote urinary iron excretion and delay progressive haemosiderosis. A splenectomy is performed in patients with gross splenomegaly and increasing need for transfusions.
Primary prevention involves screening high-risk populations, offering premarital counselling and making a prenatal diagnosis (via amniocentesis).
Acquired Haemolytic Anaemia
These anaemias can be categorised depending on the mechanism of destruction of the RBCs:
- Immune haemolytic anaemia:
- Autoimmune:
- Warm antibody – idiopathic, autoimmune diseases, infections, cancer and drugs (Coombs positive)
- Cold antibody – infection, lymphoma or autoimmune disorder
- Alloimmune – haemolytic disease, post-stem cell transplant, transfusion reactions
- Non-immune haemolytic anaemia:
- Infection e.g. malaria, haemophagocytic lymphohistiocytosis (HLH), HIV, other viruses
- Chemicals and physical causes, e.g. drugs, burns, drowning, lead poisoning
- Fragmentation, e.g. march haemoglobinuria, microangiopathic haemolytic anaemia (MAHA), cardiac haemolysis
- Acquired membrane disorder, e.g. paroxysmal nocturnal haemoglobinuria
These conditions are managed by identifying and treating the underlying cause. The mainstay of treatment in autoimmune haemolytic anaemia is steroids, given intravenously or orally. Splenectomy, rituximab, chemotherapy and immunosuppressants are second-line treatments. Some patients with acquired haemolytic anaemia may require emergency blood transfusions.
IDIOPATHIC THROMBOCYTOPENIC PURPURA (ITP)
Thrombocytopenia is diagnosed when the platelet count is <100 x109/L and patients become symptomatic with platelet counts <20 x109/L. Thus, ITP is a quantitative platelet disorder which may be acute (<1 year duration) or chronic (>1 year duration).
Aetiology
ITP is idiopathic but is associated with:
- Autoimmune disease e.g. SLE
- HIV infection
- Viral infection 2-3 weeks prior
- Lymphoproliferative disorder e.g. lymphoma, chronic lymphocytic leukaemia (CLL)
- Adverse drug reaction e.g. after immunisation
Pathophysiology
ITP platelet antibodies often cross-react, resulting in platelet sensitisation. Sensitised platelets are cleared from circulation by the macrophages of the RES, reducing the lifespan of the platelets to just a few hours.
Clinical Features
The patient may present with:
- Bleeding – mucosal bleeding (e.g. purpura), nose bleeding, menorrhagia, gum bleeding, bleeding from the urinary or GI tract
- Acute-onset of spontaneous bruising or generalised petechiae
- Intracranial bleeding (<1% of cases; can be life-threatening)
Investigations
The platelet count will be <50x109/L, and the PBS will show megakaryocytes (see related image here) and normal RBCs and white blood cells (WBCs). The bone marrow aspirate will be normal or show increased megakaryocytes and normal white and RBC precursors (the other preserved cell lines and normal lactated dehydrogenase (LDH) and uric acid help one decide between leukaemia and ITP). If the patient is >10 years old, s/he should be investigated for SLE and HIV. The platelet count should be rechecked one hour post-platelet transfusion. If it demonstrates thrombocytopaenia, peripheral platelet consumption is confirmed i.e. ITP. However, this would be an expensive diagnostic investigation and should not be routinely done.
Management
In most cases this is a self-limiting condition and does not require treatment. However, the patient should avoid drugs which may interfere with platelet function e.g. aspirin and NSAIDS.
Platelets should not be routinely transfused unless the patient is suffering from a life-threatening haemorrhage e.g. intracerebral bleeding. Even then, a transfusion should only be done in consultation with a haematologist. When there is moderate-to-severe bleeding, one should give prednisone (4 mg/kg/day) for 4 days and then repeat the FBC after 4 days.
Other therapies include:
- High-dose IV immunoglobulin (IVIg) 0.5-1 g/kg
- Alternative forms of immunosuppression e.g. azathioprine, cyclophosphamide, rituximab (anti-CD20 monoclonal antibody)
- Splenectomy (for resistant chronic ITP):
- This could be considered in children >5 years.
- The patient should receive the pneumococcal vaccine prior to splenectomy and prophylactic penicillin until the age of 18.
HAEMOPHILIA
It is an inherited coagulation disorder (see related image here) that is X-linked recessive (females are rarely affected). The partial thromboplastin time (PTT) is prolonged, while the prothrombin time (PT) and bleeding time are normal. The severity of disease is inversely proportional to factor levels.
Classification
It can be classified according to the factor deficiency:
- Haemophilia A:
- It is caused by a deficiency of factor VIII.
- Carrier females have some evidence of decreased factor VIII and lyonisation (inactivation of the second X chromosome) can result in a symptomatic female.
- Haemophilia A is most common (4-5-fold more common than haemophilia B).
30% of patients have no family history.
- Haemophilia B – caused by a deficiency of factor IX.
- Haemophilia C:
- It is caused by a deficiency of factor XI.
- It occurs equally in both sexes and is most prevalent in Jewish populations.
Haemophilia can also be classified according to the severity of the bleeding that occurs:
- Mild (occasional bleeds; 5-25% of cases)
- Moderate (less frequent bleeds post trauma or dental extraction; 1-5% of cases)
- Severe (spontaneous bleeding into joints and muscles; <1% of cases)
Clinical Features
The patient may present with a minor or major bleed.
Table 15.3: Features of Minor and Major Bleeds
| Minor Bleeds | Major Bleeds |
|---|---|
|
|
Investigations
Blood tests will show a prolonged PTT and factor levels <25% of normal.
Management
Non-Pharmacological Management
It includes:
- Genetic counselling
- Applying for a MedicAlert bracelet
- Addressing dental care
- Applying ice packs 5 min on and 10 min off in the acute bleed
- Health education for the patient (e.g. avoidance of aspirin and NSAIDs) and family on how to monitor and treat patients
- Management of knee haemarthrosis:
- Admit and avoid weight-bearing
- Immobilising the knee in slight flexion (with a backslab or plaster of Paris; POP)
- Refer to physiotherapy and occupational therapy to decide on splinting
Pharmacological Management
Bleeding episodes are treated and prevented by replacing the missing clotting factors with factor concentrate. Fresh frozen plasma (FFP) (see related image here) may be given as an alternative in an emergency situation should factor concentrate not be available. In severe haemophilia, one may consider offering 2-3-times weekly prophylactic replacement of the deficient factor. This may be continued lifelong. Patients with mild haemophilia A and some patients with Von Willebrand disease (vWD) can be treated with desmopressin (DDAVP®) prior to surgery or during bleeding episodes.
Mucous membrane bleeds can be treated with tranexamic acid (contraindicated in haematuria as may cause obstructive uropathy). Surgical and dental procedures should be done under factor cover and a multidisciplinary approach is mandatory. The patient may become refractory to replacement of coagulation factor, as a result of developing high levels of circulating inhibitors. These may be managed through inhibitor eradication by immune tolerance induction (ITI) or the use of bypass agents e.g. anti-inhibitor coagulant complex (FEIBA®) or emicizumab.
DISSEMINATED INTRAVASCULAR COAGULOPATHY (DIC)
It is an acquired condition characterised by simultaneous bleeding diathesis and microvascular thrombosis.
Aetiology
DIC may be caused by:
- Septicaemia and shock
- Trauma e.g. burns, head injury, post-operative states
- Malignancy e.g. promyelocytic leukaemia
- Perinatal complications e.g. birth asphyxia, necrotising enterocolitis
Viraemia
DIC is also associated with Kaposiform haemangio-endothelioma (Kasabach-Merritt syndrome).
Pathophysiology
The inciting event induces endothelial damage and activates the coagulation cascade, which consumes clotting factors and platelets and inhibits natural anticoagulants and fibrinolysis. This process continues uncontrollably, resulting in DIC. Depending on the extent of the thrombosis, end-organ failure may develop (kidney, liver, lung, brain, adrenals, heart or extremities).
Clinical Features and Investigations
Haemorrhage is the most prominent clinical feature with the most common sites of bleeding being the skin (purpura), venipuncture sites, GIT, lungs (intrapulmonary bleeding) or brain (intraventricular haemorrhages). The patient will also have abnormal coagulation studies (PTT, low fibrinogen levels and elevated D-dimer levels).
Management
One must treat the underlying cause and give supportive care, including FFP and cryoprecipitate transfusions. Blood and platelet transfusions may also be given, as 319 needed. Anticoagulation is generally not indicated unless there is significant thrombosis e.g. purpura fulminans.
VON WILLEBRAND DISEASE (vWD)
It is the most common congenital bleeding disorder. Von Willebrand factor (vWF) is a large multimeric protein that is essential for primary haemostasis and is the carrier for factor VIII.
Low levels of vWF affect up to 1% of the population, however individuals are rarely symptomatic. vWD may be autosomal dominant or recessive.
Pathophysiology
The deficiency of vWF (due to a quantitative deficiency or qualitative deficiency i.e. abnormal vWF) results in lower platelet adhesiveness and an impairment of primary haemostasis.
Clinical Features
The patient may present with:
- Family history or significant personal history of bleeding
- Recurrent epistaxis and mucosal bleeding e.g. easy gum bleeding, easy bruising, prolonged bleeding from injuries
- Heavy menstrual bleeding at the onset of menarche in girls
Investigations
Blood tests will show
- Prolonged bleeding time
- Prolonged PTT
- screening tests – may be normal
- Decreased vWF antigen
- Decreased ristocetin cofactor and/or collagen binding activity
- Decreased factor VIII coagulant activity
Management
Non-pharmacological management includes first aid (especially for epistaxis) and the application of pressure to bleeding areas e.g. using tranexamic acid-impregnated gauze to apply pressure for tooth socket bleeds.
Pharmacological management includes
- For mild bleeds – IV desmopressin (maximum 3 doses)
- For severe bleeds – factor VIII or vWF concentrate
- For mucous membrane bleeds – tranexamic acid
- For menorrhagia – low-dose combined oral contraceptive
ONCOLOGY
At the link is a mnemonic for the early paediatric oncological signs by the Childhood Cancer Foundation of South Africa (CHOC), based on Saint Siluan’s early warning signs of childhood cancer (see related link here).
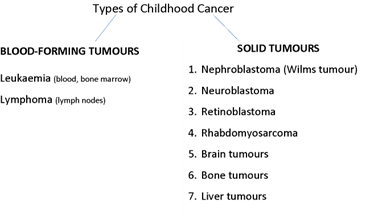
Classification
Childhood cancers may be classified as blood-forming tumours and solid tumours; see related image to lymph nodes here.
LEUKAEMIA
Acute leukaemia is the most common cancer in childhood. The leukaemia can either have a lymphoid- or myeloid-precursor origin – ALL or acute myeloid leukaemia (AML), respectively (see related image here). ALL can either have a B-cell lineage (B-ALL) or T-cell lineage (T-ALL).
Pathophysiology
Leukaemia is caused by uncontrolled replication of a single, immature, haematopoietic WBC with arrested or abnormal differentiation. It is, therefore, a clonal disease. Proliferation of the cells leads to the infiltration of bone marrow by blasts and replacement of normal haematopoietic cells. The abnormal cells circulate in the bloodstream and infiltrate other tissues.
Clinical Features
Leukaemia peaks in children aged 1-5 years and is slightly more common in males. The child may present with an acute history (days to weeks) of:
- Pallor
- Fatigue, lethargy and irritability
- Headache
- Generalised lymphadenopathy
- Hepatosplenomegaly
- Bone pain
- Fever
- Bleeding (epistaxis or gingival), bruising and petechiae
- Chloroma
- Gingival hypertrophy
- Testicular enlargement
- Focal neurological signs and/or seizures (if there is CNS involvement)
- Dyspnoea, orthopnoea and/or tachypnoea (if there is a mediastinal mass)
Investigations
They should include:
- FBC – will show:
- High, normal or low WCC
- Normochromic, normocytic anaemia
- Thrombocytopaenia
- RFTs – will show:
- High LDH
- High urate
- PBS – will show blasts
- Bone marrow aspirate and trephine biopsy (BMAT)
- A sample should be sent for morphology, immunophenotyping (flow cytometry) and cytogenetics.
- CXR – to exclude a mediastinal mass
- Abdominal ultrasound scan (USS) – to demonstrate hepatosplenomegaly
- LP – to demonstrate the presence of blasts and confirm CNS disease
Management
Treatment is adapted based on prognostic factors. One must treat infections and manage bleeding and tumour lysis syndrome (TLS).Steroids (prednisone and dexamethasone) and chemotherapeutic agents (vincristine, daunorubicin and asparaginase) are the mainstays of treatment. Intrathecal chemotherapeutic agents may also be used (methotrexate is used to prevent or treat CNS disease).
Maintenance chemotherapy, with mercaptopurine, methotrexate, vincristine and dexamethasone, is continued for two years in girls and three years in boys. A bone marrow transplant is indicated for high-risk or relapsed leukaemia (see related image here).
SOLID TUMOURS
LYMPHOMA
Paediatric lymphomas can be divided into Hodgkin’s lymphomas (HLs) and non-Hodgkin’s lymphomas (NHLs). HLs can be further subdivided into classical and nodular, lymphocyte-predominant HLs. NHLs can either have a T-cell origin (e.g. 323 anaplastic large cell lymphoma) or B-cell origin (e.g. Burkitt lymphoma, diffuse, large B-cell lymphoma).
Clinical Features
The child will present with:
- Lymphadenopathy:
- Painless nodal enlargement
- Often chronic lymphadenopathy in HLs
- Presence of B symptoms:
- Anorexia and weight loss
- Fever
- Drenching night sweats
Children with Burkitt’s lymphoma may have:
- Jaw mass (endemic Burkitt’s lymphoma)
- Abdominal mass (sporadic Burkitt’s lymphoma)
- Acute abdomen with intussusception and bowel obstruction or acute bowel perforation
- Ascites and/or kidney infiltration
- Disseminated disease (bone marrow and/or CNS involvement)
Patients with HLs may present with:
- Painless lymphadenopathy (usually spreads to contiguous nodes in a predictable sequence)
- Generalised pruritus (may indicate the presence of immune haemolytic anaemia)
- Mediastinal lymphadenopathy (may manifest as airway compression or superior vena cava obstruction)
Investigations
They should include:
- Fine needle aspiration biopsy (FNAB) and/or excision biopsy for histology:
- HLs will show Reed-Sternberg cells on a background of other inflammatory cells.
- Burkitt’s lymphoma will have a ‘starry sky’ appearance and a high Ki-67 proliferation index (indicating a high mitotic rate).
- Chest X-ray (to exclude a mediastinal mass)
- Abdominal USS (to exclude an abdominal mass and/or lymphadenopathy)
- Bone marrow aspirate (to investigate for bone marrow involvement)
- LP (to investigate for CNS involvement)
NHLs will then be staged using the St Jude/Murphy staging system and HLs using the Ann Arbor staging system.
Management
Table 15.4: Management of Non-Hodgkin’s and Hodgkin’s Lymphomas
| NHLs | HLs |
|---|---|
|
|
WILMS TUMOUR (NEPHROBLASTOMA)
It is the most common renal malignancy in children and remains one of the most common malignancies in Africa. It usually occurs in children <5 years old and is more common in females. In 5-10% of cases, both kidneys are affected. Wilms tumour is associated with genetic conditions in which there is a WT-1 gene deletion or mutation, such as:
- WAGR syndrome (Wilms tumour, aniridia, genito-urinary abnormalities and mental retardation)
- Beckwith-Wiedemann syndrome (characterised by macroglossia, macrosomia, omphalocele and hemi-hypertrophy)
- Denys-Drash syndrome (characterised by diffuse mesangial sclerosis leading to progressive renal failure, male pseudohermaphroditism and Wilms tumour)
Clinical Features
They may include:
- Incidental finding of an abdominal mass by the parent or doctor
- Associated congenital anomalies (present in 20% of cases)
- Aniridia
- Hemihypertrophy
- Urogenital anomalies
- Fever
- Abdominal pain
- Haematuria
- Hypertension
- Anaemia (may be caused by intratumoural bleeding or rupture)
- Acquired vWD and associated bleeding
- Severe malnutrition (may be present in advanced disease)
5% of affected children present with bilateral tumours.
Investigations
One should order:
- FBC
- RFTs
- LFTs
- Coagulation screen
- Urinalysis:
- It is done to look for blood and protein and to look for urine catecholamines to exclude neuroblastomas.
- Ultrasound:
- This non-invasive investigation is done to identify the tumour site and characteristics, bilateral or hepatic involvement, nephrogenic rests, abdominal lymphadenopathy, and renal vein or IVC thrombosis.
- MRI abdomen (to better delineate the renal mass)
- Chest X-ray or CT lung (to detect lung metastases)
- FNAB:
- It is done to confirm the diagnosis and determine the prognosis.
- Focal or diffuse anaplasia is associated with a worse prognosis and will require radiotherapy.
Management
Treatment is determined by the stage of the disease but multimodal therapy is often required (chemotherapy, surgery and radiotherapy).
NEUROBLASTOMA
It is the most common extracranial solid tumour in children. The median age of onset is 2 years. The neoplastic cells are derived from primordial neural crest cells which develop into sympathetic ganglia and the adrenal medulla. The abdomen is the most frequent location for this cancer (suprarenal mass) but the disease can manifest anywhere along the sympathetic chain (from neck to pelvis). There is great heterogeneity in this disease, with some patients undergoing spontaneous regression and others having resistant and aggressive disease.
Clinical Features
The patient will have general clinical features (fever, anorexia, lethargy, pallor, weight loss and irritability) and organ-specific clinical features.
- Abdominal disease – abdominal mass associated with pain, anorexia, vomiting or constipation
- Thoracic disease – posterior mediastinal mass associated with dyspnoea, orthopnoea or dysphagia
- Head and neck – Horner’s syndrome
- Pelvic – difficulty micturating and defecating
- Paraspinal disease (extension to the neural foramen) – spinal cord compression with associated symptoms dependent on the level of compression e.g. pain, paralysis and issues with bladder and bowel function
- Disseminated disease (common) – swellings on the skull, proptosis and raccoon eyes (blue periorbital discolouration), skin metastasis (blueberry muffin rash), painful skeletal metastasis, distant lymph node metastasis or liver metastasis
- Paraneoplastic phenomena – chronic diarrhoea, opsoclonus-myoclonus ataxia syndrome (dancing eyes) and hypertension
Investigations
They should include:
- FBC (to assess for bone marrow infiltration)
- Tumour markers (raised LDH and ferritin)
- Abdominal USS (to assess for an abdominal mass)
- Chest X-ray (to assess for a mediastinal or paraspinal mass)
- Bone scan (to look for skeletal metastasis)
- BMAT (to look for BM involvement)
- Biopsy (to assess the histology and biology of the tumour; MYCN amplification is associated with a very poor prognosis)
- Urine catecholamines (homovanillic acid and vanillylmandelic acid)
- CT/MRI (to better delineate an abdominal mass from a paraspinal mass)
- Metaiodobenzylguanidine (MIBG) scan (to assess uptake in the primary mass and assess for skeletal deposits)
Management
A multimodal approach is used, with chemotherapy, surgery and radiotherapy all being used. An autologous stem cell transplantation may also be performed. Maintenance therapy is with anti-GD2 immunotherapy and cis-retinoic acid.
RETINOBLASTOMA
It is the most common intraocular tumour in childhood and occurs in children <5 years old. The tumour can be unilateral or bilateral, unifocal or multifocal, and sporadic or hereditary. Bilateral, multifocal, hereditary retinoblastoma is associated with RB1 gene mutations or deletions. In hereditary retinoblastoma there is an increased risk of developing secondary malignancies, including the development of an asynchronous brain tumour (trilateral RB) (see related image here).
Genetic counselling is indicated in all patients. Patients should be screened for the development of retinoblastoma in the contralateral eye and for the development of an asynchronous brain tumour. Siblings and future offspring should be screened for retinoblastoma.
Clinical Features
Patients may present with leukocoria (absent red reflex on flash photography) and/or strabismus early in the disease process. Children with locally advanced disease present with a painful, red eye, proptosis and/or glaucoma. In metastatic disease, there is extension to the optic nerve (causing blindness) or cerebrum (causing localising neurological signs, seizures or raised intracranial pressure).
A thorough ophthalmological examination should be performed. One may need to use a RetCam® or examine the child under anaesthesia.
Investigations
They should include:
- USS of the eye (to look for a hyperechoic mass and calcifications)
- CT/MRI of brain and orbit (to assess for extension to the optic nerve, orbit or cerebrum)
- Bone marrow aspiration (to assess for BM involvement)
- LP (to assess for CNS involvement)
Management
A well-coordinated, multidisciplinary team approach is required. One’s priorities are to preserve life, preserve vision and preserve the eye. Treatment is dependent on the stage and the extent of the disease.
For small tumours, one should consider local therapies (photocoagulation, cryotherapy, intralesional or intra-arterial chemotherapy and/or brachytherapy). For larger tumours which are limited to the eye, enucleation is the intervention of choice. For advanced or metastatic disease, chemotherapy and external beam radiotherapy are required. CNS disease may require intrathecal chemotherapy and craniospinal irradiation.
RHABDOMYOSARCOMA
It is the most common soft-tissue sarcoma of childhood, and is generally diagnosed ~5 years old. Approximately half of the tumours occur in the head and neck region (orbit, nasopharynx, middle ear or face), a quarter in the genitourinary system (bladder, prostate, vagina, uterus or paratesticular space) and the remaining quarter in the extremities, trunk, or retroperitoneum. Metastases occur early and are to regional lymph nodes, lungs, bones, and bone marrow.
Clinical Presentation
Tumours of the ear, nose, bladder, uterus or vagina may present as polypoid lesions, which cause obstruction or secrete a bloodstained, offensive discharge. On the trunk and extremities, the tumours present as soft-tissue masses (may be tender and easily confused with an acute abscess). Orbital swelling may be mistaken for a retinoblastoma, neuroblastoma or Burkitt’s lymphoma. The brain and cerebrospinal fluid may be infiltrated with parameningeal tumours.
Investigations
One should perform a FNAB or incisional/excisional biopsy of the lesion. The extent of metastatic spread must also be investigated (CT, CXR, PET scan, etc.).
Management
A multimodal approach must be followed (chemotherapy, surgical resection and radiotherapy). Primary total surgical resection followed by chemotherapy or chemotherapy followed by complete resection of residual tumour offers the best chance of cure. Radiotherapy must be done at tumour sites where surgical removal has not produced a microscopically complete tumour resection.
Prognosis
Children with limited disease which has been completely excised have a good prognosis. Those with advanced disease, with incomplete resection or who are <1 year or >10 years old have a worse prognosis.